Introduction¶
Overview¶
The Tentacle framework is developed to enable researchers to leverage high-performance computer (HPC) systems to quantify genes in large metagenomic data sets.
Tentacle provides a way to distribute resource intensive mapping tasks, such as mapping large numbers of metagenomic samples to a reference, in order to reduce the time required to analyze large data sets. Tentacle makes it possible to define what mapper and mapping criteria to use, and computes the coverage of all annotated areas of the reference sequence(s).
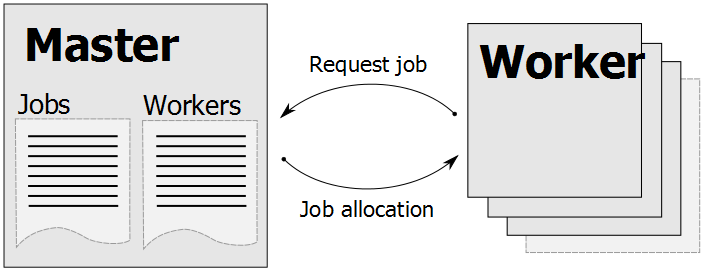
Tentacle uses a master-worker scheme that has Master process that maintains a list of mapping jobs that should be run. The Worker processes (normally running on computer nodes in a cluster) connects to the Master process and asks for a mapping job to run.
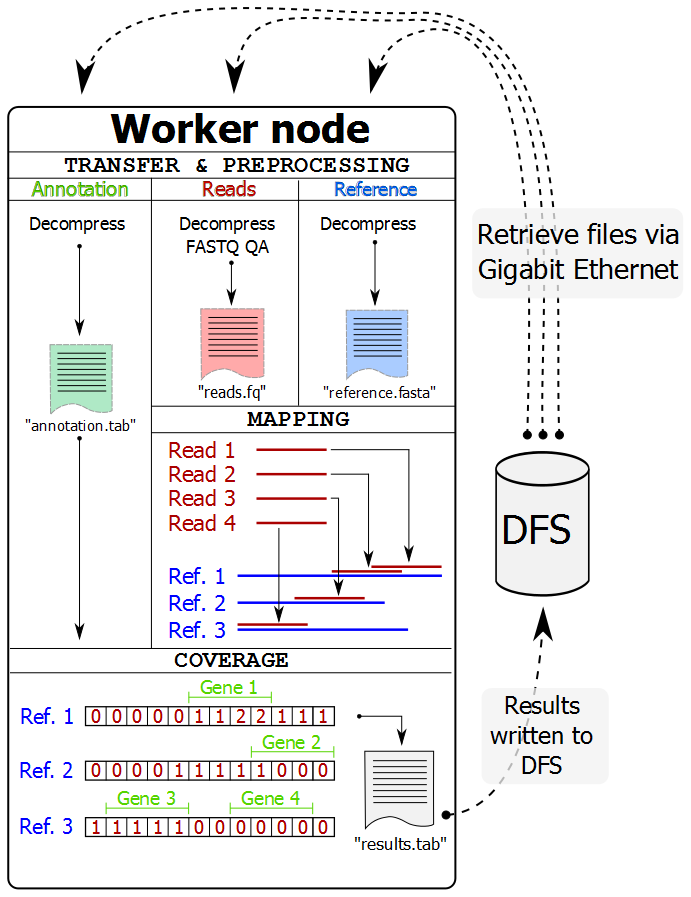
The Worker processes (normally on a separate computer node in a cluster) perform all their work without interaction with the Master process. They retrieve the files via network from a distributed file system (DFS), perform preprocessing, mapping, and coverage calculations and then write the results back to the distributed file system. After completing a job, the Worker process requests a new one from the Master process.
More information available on how Tentacle computes coverage is available in section Coverage. Further information on the implementation will also be published.
Use examples¶
Tentacle is well suited for the following tasks:
- Map samples of metagenomic reads back to previously annotated contigs to determine the presence and abundance of certain genes. Check out TUTORIAL 1. Mapping reads to contigs (pBLAT).
- Map metagenomic reads to a reference database of e.g. antibiotic resistance genes. Check out TUTORIAL 2. Mapping nucleotide reads to amino acid database (USEARCH).
Abstract¶
Metagenomics is the study of microorganisms by sequencing of random DNA fragments from microbial communities. Since no cultivation of individual organisms is required, metagenomics can be used to analyze the large proportion of microorganisms that are hard or impossible to grow in laboratories. Metagenomics therefore holds great promise for understanding complex microbial communities and their interactions with their respective environments. The analysis of metagenomes in human gut, oral cavities and on our skin can, for example, provide information about what microorganisms are present and their effects on human health. The introduction of high-throughput DNA sequencing has significantly increased the size of the metagenomes which today can contain trillions of nucleotides (terabases) from one single sample. Current methods are however not designed with these large amounts of data in mind and are consequently forced to significantly reduce the sensitivity to achieve acceptable computational times. We have therefore developed a new method for distributed gene quantification in metagenomes. In contrast to many existing methods that are designed for single-computer systems our methods can be run on computer clusters and grids. Through efficient data dissemination and state-of-the-art sequence alignment algorithms the framework can rapidly and accurately estimate the gene abundance in very large metagenomes while still maintaining a high sensitivity. The output of our framework is adapted for further statistical analysis, for example comparative metagenomics to identify both quantitative and qualitative differences between metagenomes. Our method is shown to scale well with the number of available compute nodes and provides a flexible way to optimally utilize the available compute resources. It provides fast and sensitive analysis of terabase size metagenomes and thus enables analysis of studies of microbial communities at a resolution and sensitivity previously not feasible.
Copyright and license¶
Tentacle was developed at Chalmers University of Technology and is Copyright Fredrik Boulund, Anders Sjögren, and Erik Kristiansson. The code is freely available for use, re-use, and modification under the GPL (see Citing Tentacle).